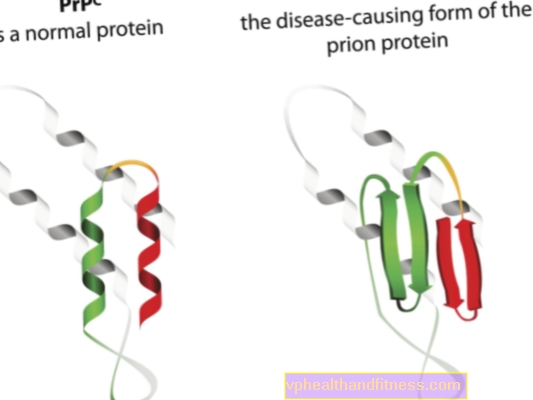
Губчасті енцефалопатії (пріонні хвороби) - це ті захворювання, при яких у розвитку беруть участь патологічні форми пріонних білків. Ми знаємо все більше і більше про пріонні хвороби, але ключові аспекти досі невідомі - в даний час медицина не має засобів для лікування пацієнтів від цих хвороб.
Губчасті енцефалопатії, тобто пріонні хвороби, можуть розвиватися протягом життя, тоді як інші виникають внаслідок спадкових генних мутацій, присутніх від народження. У цій групі існує декілька істот, що трапляються у людей, наприклад, хвороба Крейцфельдта-Якоба або фатальне сімейне безсоння.
Пріонові хвороби дуже давно були загадковими. На відміну від інших патогенних мікроорганізмів, таких як бактерії, віруси або гриби, вони не містять нуклеїнової кислоти - пріони утворюються лише з білків. Теорію пріонних хвороб відкрив С. Прузінер, це відкриття було високо оцінено науковим співтовариством - в 1997 році дослідник отримав Нобелівську премію з медицини. Незважаючи на те, що минуло порівняно багато років з часу народження концепції пріона, деякі вчені досі вважають, що вона неповна, і досліджують далі природу цих станів - деякі з факторів, відповідальних за губчасті енцефалопатії, зараз підтверджені.
Пріонові хвороби: причини
Етіологія пріонних хвороб пов’язана з перетворенням нормальних пріонних білків у патогенні, патогенні форми. Пріони - це білкові молекули, які містяться в організмі кожної людини. Їх функція ще не зовсім зрозуміла, але відомо, що в нормальних умовах пріонні білки не шкодять організму. Але коли пріони змінюють свою структуру і стають патогенними частинками, розвивається одна з кількох губчастих енцефалопатій. Пріони, що зустрічаються в організмі в природі, називаються PRPC, тоді як аномальні форми - PRPSC. Останні становлять серйозну проблему не тільки тому, що вони можуть накопичуватися в нервовій тканині у вигляді відкладень і породжувати її пошкодження, але й тому, що вони мають здатність перетворювати нормальні пріони в деформовану форму (простіше кажучи, PRPSC може "заражати" нормальні білки з їх патогенним потенціалом).
Читайте також: Хвороба Хантінгтона (хорея Хантінгтона): причини, симптоми, лікування Тремтіння м’язів - причини. Що означає тремтіння м’язів? Хвороби, які вбивають найшвидше: ШОК, ЕБОЛА, ПРОКЛЯ, НАПАД, НЕЗАЛЕЖНІ [ГАЙЛ ...В основному є 3 причини губчастої енцефалопатії:
- епізодичні (патогенна мутація виникає в соматичних клітинах, це відбувається протягом життя пацієнта),
- сім'я (внаслідок тягаря мутацій, успадкованих від батьків),
- пасажоподібні (пов’язані з введенням патогенних пріонів в організм людини, наприклад, через препарати гормону росту, забруднені цими частинками, або трансплантацію рогівки від людини, яка страждає на якусь губчасту енцефалопатію).
Губчасті енцефалопатії: хвороба Крейцфельдта-Якоба
Хвороба Крейтцфельдта-Якоба (CJD) була вперше описана на початку 1920-х років. Існує 4 типи захворювання:
- спорадичний CJD (найпоширеніший, що становить до 9/10 всіх випадків CJD)
- рідне місто CJD
- ремінь CJD
- варіант CJD
Клінічна картина при різних варіантах хвороби Кройцфельдта-Якоба може бути різною. Найбільш типовими недугами в процесі цієї групи губчастих енцефалопатій є:
- розлади деменції (включаючи поступове погіршення пам’яті, уваги та концентрації уваги)
- міоклонія (мимовільні рухи, як раптові поштовхи м’язів)
- дисфункція мозочка (проявляється, наприклад, порушеннями рівноваги)
- затуманений зір
- пірамідні та екстрапірамідні симптоми
Під час варіантів CJD можуть також з’являтися психічні розлади (наприклад, тривога, пригнічений настрій), біль та інші мимовільні рухи, крім згаданих вище.
Прогноз хвороби Крейтцфельдта-Якоба поганий - наприклад, у пацієнтів із епізодичною ХЖН в середньому проходить від чотирьох до п’яти місяців від появи симптомів захворювання до смерті.
Губчасті енцефалопатії: синдром Герстмана-Штрауслера-Шейнкера
Синдром Герстмана-Штрауслера-Шейнкера (GSS), як правило, протікає в сім'ях і викликаний спадковою мутацією гена PRNP. Вважається, що це найбільш повільно прогресуюча губчаста енцефалопатія. Команда GSS включає:
- спиноцеребелярна атаксія
- дизартрія
- деменція
- порушення ковтання
- ністагм
- підвищена напруга м’язів
Пацієнти з діагнозом GSS мають різну кількість часу, і у деяких пацієнтів смерть настає понад 10 років після початку.
Губчасті енцефалопатії: фамільне сімейне безсоння
Фатальна сімейна безсоння - пріонна хвороба, спричинена мутаціями гена PRNP. Хвороба надзвичайно рідкісна і до цього часу діагностована у 28 сім’ях по всьому світу. Під час фатальної сімейної безсоння першим симптомом є нездатність спати. Ця проблема призводить до тривожних розладів і у пацієнта виникають галюцинації. Ефектом постійної відсутності нічного відпочинку є порушення у функціонуванні вегетативної системи (включаючи зміни в роботі серця, потовиділення та розлади травної системи), спостерігається також поступове зменшення маси тіла. На більш запущених стадіях фатальної сімейної безсоння з’являються гормональні порушення, а симптоми деменції з’являються в процесі захворювання.
Прогноз сімейної безсоння, як і інших губчастих енцефалопатій, поганий: пацієнти зазвичай помирають протягом трьох років від початку захворювання.
Губчасті енцефалопатії: пріонопатія зі змінною сприйнятливістю до протеази
Виникнення обговорюваних губчастих енцефалопатій головним чином пов’язане з мутаціями гена PRNP. Однак ці мутації стосуються різних кодонів цього гена, і тому виділяють кілька різних пріонних хвороб. Порівняно недавно описана (у 2008 р.) Одиниця - пріонанопатія зі змінною сприйнятливістю до протеази. Люди, які страждають цією хворобою, мають мутації до трьох кодонів гена PRNP.
При пріонопатії зі змінною сприйнятливістю до протеази пацієнти відчувають:
- когнітивні порушення
- надзвичайна тяжкість психічних розладів: це може бути ейфорія та збудження, але також значна апатія
- дизартрія
- афазія (мовні розлади)
Середня тривалість захворювання при цій пріонопатії становить менше 4 років.
Губчасті енцефалопатії: куру
Зараз Куру вважають хворобою, яка практично вже не існує - її виявили у представників племен з Папуа-Нової Гвінеї, які практикували канібалістичну поведінку. Домінуючим симптомом цієї губчастої енцефалопатії є прогресуюча атаксія мозочка. Це може супроводжуватися мимовільними рухами (переважно у вигляді хореї, тремтіння та атетозу), а також нетриманням сечі та фекалій. Пацієнти на куру також відчувають значні зміни настрою, у них формуються примітивні рефлекси (наприклад, смоктання). Досить характерною проблемою у випадку цієї пріонової хвороби є вимушені напади плачу або сміху - через останні явища, куру іноді називають "смех смертю".
Губчасті енцефалопатії: діагностика
Запідозрити пріонні захворювання можна на основі симптомів пацієнта. Однак вони досить неспецифічні, оскільки можуть з’являтися і в процесі ряду інших захворювань, які не пов’язані з пріонами. З цієї причини при діагностиці губчастих енцефалопатій також використовуються:
- візуалізаційні тести (наприклад, магнітно-резонансна томографія, що дозволяє виявити зміни, пов’язані з дегенерацією мозку пріонними білками),
- лабораторні тести (такі як оцінка концентрації білка в лікворі, наприклад MAP-тау, білки S-100 або 14-3-3),
- генетичні тести (для виявлення наявності мутацій у пацієнта),
- імуногістохімічні тести (з використанням антитіл до прионних білків).
Діагноз також може бути підтверджений розтином мозку, при якому можна виявити зміни, характерні для губчастих енцефалопатій. Це можуть бути губчасті ураження, по-різному розподілені та з різною структурою (залежно від конкретної сутності захворювання) амілоїдними бляшками та дефектами нейронів.
Губчасті енцефалопатії: лікування
В даний час пріонні хвороби невиліковні - незважаючи на численні дослідження, які тривають вже багато років, медицина досі не має препаратів, які могли б уповільнити або повністю загальмувати їх розвиток. У пацієнтів із губчастими енцефалопатіями застосовується симптоматичне лікування, яке спрямоване на пом’якшення інтенсивності симптомів та максимально поліпшення якості їх життя.
Однак робота з лікування губчастих енцефалопатій все ще триває. Вчені намагаються використовувати різні методи - перший приклад - це генна терапія. Вони впливали б на нуклеїнові кислоти та мутації, що присутні в їх структурі - метою застосування генної терапії було б нейтралізувати помилки в генетичному коді. Інший підхід лежить в основі імунної терапії - триває робота зі створення антитіл, роль яких полягала б у виведенні патогенних пріонів. Іншим методом, який бачить потенціал боротьби з губчастими енцефалопатіями, є лікування із застосуванням синтезованих молекул білка, які, потрапляючи в організм пацієнта, нейтралізують патологічні білки.
Рекомендована стаття:
Енцефалопатії - причини, типи та симптоми









-trzy-gatunki-rne-choroby.jpg)